‘Er is geen erfelijke link. Hij heeft brute pech: een fout in de celdeling’: deze families leven met de chromosomale afwijking 22q11

Op 22 november is het internationale bewustwordingsdag van het 22q11-deletiesyndroom, na downsyndroom de meest voorkomende chromosomale afwijking. Want wat het precies is en hoe het zich uit, is nog veel te weinig bekend.
Het 22q11-deletiesyndroom is na downsyndroom de meest voorkomende chromosomale afwijking. In Vlaanderen worden elk jaar 15 tot 20 nieuwe diagnoses gesteld, en toch hebben veel mensen – zelfs artsen – nog nooit van de aandoening gehoord. De ziekte ontstaat door een ontbrekend stukje DNA op chromosoom 22, en is meestal niet erfelijk. De symptomen zijn enorm divers, en de ernst ervan varieert van patiënt tot patiënt. Meest voorkomend zijn hartproblemen, een defect van het gehemelte, leer- en gedragsproblemen en een zwak immuunsysteem. Op 22 november is het internationale bewustwordingsdag, en ook deze mensen proberen 22q11 uit de onbekendheid te halen.
Tine Maesen (40) en Lotta (7,5)

“Toen ik 32 weken zwanger was, had ik te veel vruchtwater en een verkorte baarmoederhals. Lotta had een groeiachterstand en een hartafwijking. De NIP-test (prenatale test om diverse afwijkingen op te sporen, SP) was toen, net als vandaag, nog niet gevoelig genoeg om 22q11 op te sporen. Een vruchtwaterpunctie was de enige optie, maar die zou het risico op een vroege bevalling verhogen. De dokters spraken in bedekte termen: “Het zou kunnen dat je kindje 22q11 heeft, maar we kunnen nog geen uitsluitsel geven. Wat je ook doet: begin het niet te googelen.” Uiteraard doe je dat wel. Bij elke muisklik stortte onze wereld wat verder in. Die laatste weken waren zenuwslopend, bijna ondraaglijk. We wilden gewoon onze dochter zien.
“Na de positieve test probeerden de dokters ons gerust te stellen: ‘De symptomen kunnen ook meevallen. Het kan zelfs zijn dat ze er niets van merkt.’ Al snel bleek dat Lotta zich aan de zwaardere kant van het 22q11-spectrum bevindt. Ze had strakke spieren, kon pas heel laat zitten en staan, en lachte veel later dan andere baby’s. Op aanraden van de dokters hadden we de diagnose verzwegen, zelfs voor onze dichte familie. Als ze weinig symptomen had, moest ze niet met die stempel leven. Maar na een tijdje merkte iedereen dat er iets niet klopte.
“Vanaf 12 weken kreeg ze kinesitherapie. Toen ze zes maanden was, startten we met logopedie om haar te helpen eten, en later om beter te spreken. Ze kreeg een oogcorrectie omdat haar hersenen visuele prikkels verkeerd verwerkten. Lotta keek scheel naar buiten waardoor ze een heel breed gezichtsveld had. Ze kon een toren maken met blokken die links achter haar stonden.
“Vandaag heeft ze een emotionele leeftijd van een kind van vier, maar ze begint nu wel te beseffen dat een bezoek aan het ziekenhuis vaak niet vrijblijvend is. Bij de oorarts moesten we haar onlangs met vier vasthouden. Als ze uit narcose komt, is ze een wild beestje, helemaal in paniek. Het breekt mijn hart dat ik haar als moeder niet altijd kan beschermen.
“Lotta is een ‘de novo’. Mijn man Robin en ik zijn allebei geen dragers. Maar zelfs zonder die erfelijke belasting hebben we getwijfeld om opnieuw zwanger te worden. Bij Wess, onze zoon die aan wiegendood stierf toen hij drie maanden was, en bij onze dochter Essie kozen we telkens voor een vruchtwaterpunctie. Het is niet de bedoeling dat Essie later voor Lotta zal zorgen, maar we wilden niet dat ze een enig kind bleef. Ze zal waarschijnlijk nooit alleen kunnen wonen of een normale job hebben. Dat vind ik het moeilijkste aan dat syndroom. Niet de constante zorg, maar het besef dat er een dag komt waarop wij niet meer voor haar kunnen zorgen. Ik wil niet dat ze in een instelling terechtkomt als wij er niet meer zijn. We hopen zelf ooit te kunnen investeren in voorziening waar ze zelfstandig kan wonen.
“Dit leest als een loodzwaar boek, en dat is het ook, maar Lotta is vooral een ontzettend lief doorzettertje. Wij zien haar als onze Lotta, niet als een kind met een beperking. Eigenlijk hebben we ontzettend veel ‘geluk’ gehad. Omdat de aandoening zo weinig bekend is, moeten sommige mensen tien jaar naar de juiste diagnose zoeken. Wij werden meteen doorverwezen naar een kinderarts en orthopedagoog met tonnen ervaring. Maar eigenlijk zou je hier geen geluk mogen hebben. De juiste kennis en ondersteuning zou voor iedereen toegankelijk moeten zijn.”
Mieke Vanwezer (47) en Femke (7)
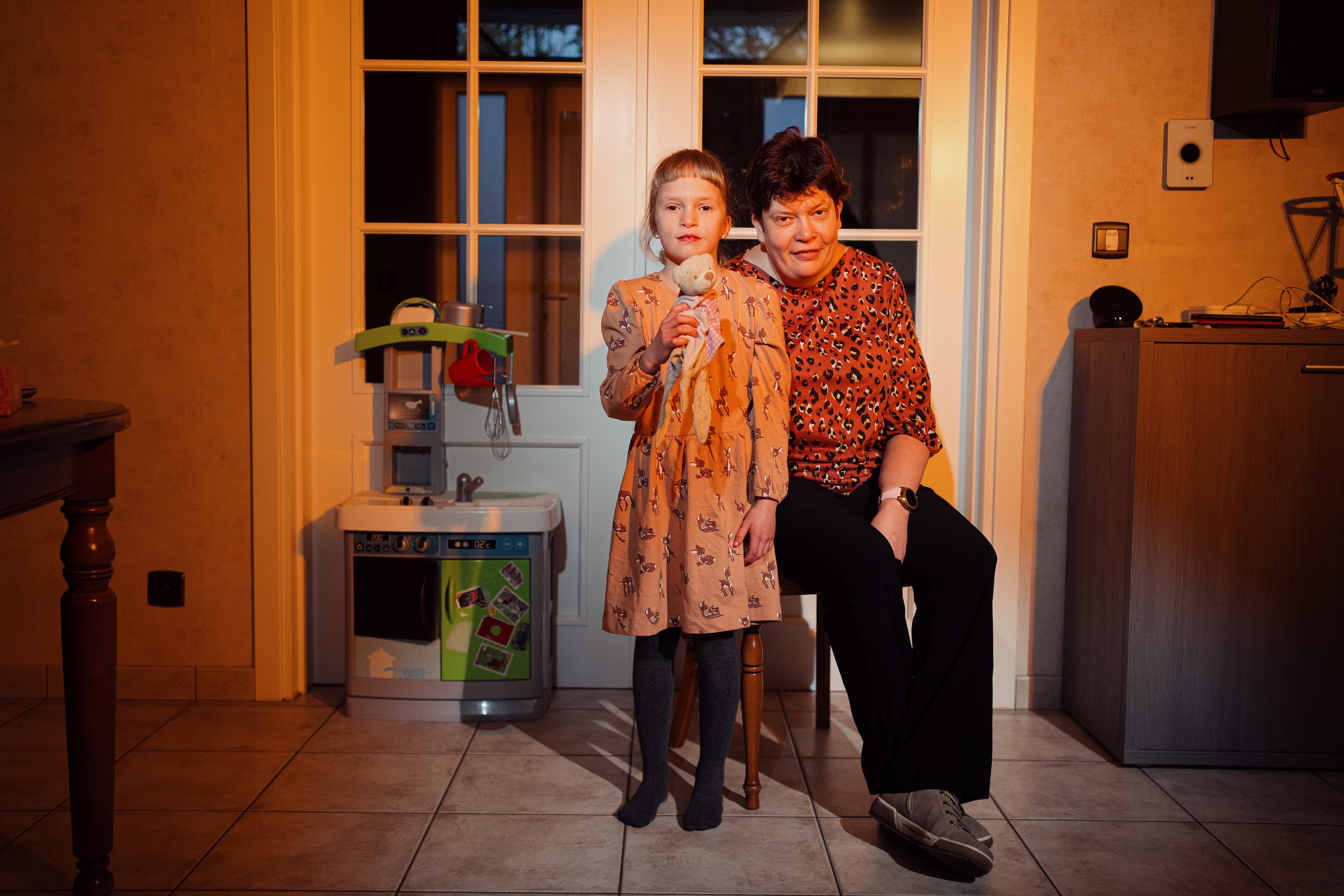
“Begin jaren negentig werd mijn jongste zus geopereerd aan een gaatje tussen haar hartkamers. De dokters hadden een vermoeden dat 22q11 er iets mee te maken had, dus werden we getest: mijn twee zussen, onze moeder en ik. We testten allemaal positief, behalve de middelste zus. Het was meteen een verklaring voor de schizofrenie en psychotische episodes van mijn moeder. Ze werd verscheidene keren opgenomen in een psychiatrisch ziekenhuis, en als ze thuis was, zat ze als een plant in de zetel, verdoofd door pillen. Ik herinner me nog hoe ik rond haar zetel poetste en zij alleen maar voor zich uit bleef staren. Een gesprek met haar aangaan, was moeilijk.
“Ze bood geen geborgenheid, integendeel. Praten deden we als gezin sowieso weinig. We hebben die diagnose zelfs nooit op tafel gesmeten. Het was elk voor zich. Hoofd naar beneden en voortdoen. Ik moest als oudste dochter mee het huishouden draaiende houden. Mijn moeder was afwezig en mijn vader had het druk met lesgeven. Ik heb, denk ik, een gewone jeugd gehad. Maar ik heb me wel vaak alleen gevoeld.
“Vlak na de diagnose zeiden de dokters dat ik eerst op raadpleging moest komen als ik ooit kinderen wou. Als je de ziekte zelf hebt, heb je één kans op de twee om ze door te geven. Ze gaven die diagnose en een vaag toekomstperspectief, maar boden me geen enkele vorm van opvolging of begeleiding. Toen ik zeventien jaar later mijn eerste kind kreeg, was 22q11 een schim uit het verleden. Pas toen Femke zeven jaar geleden geboren werd, kwam alles terug. De kinderarts ontdekte een hartgeruis. Uit de echocardiogram bleek dat ook Femke een gaatje had tussen haar hartkamers. Het was bijna een centimeter groot. Door haar zwakke hart had ze zodanig weinig kracht dat aanhappen niet lukte. We schakelden over naar flessenvoeding, maar ze deed er een uur over om een flesje uit te drinken. Op zes weken werd ze geopereerd. Ze is nu zeven, maar eigenlijk is ze een kind van vier. Ze zit in het buitengewoon onderwijs. Elk jaar heeft ze wel een ziekenhuisopname met een dubbele longontsteking. Bovendien hoort ze slecht, waardoor haar taal laat op gang kwam. Haar grote broer, die hoogbegaafd is, bekommert zich als de beste om zijn kleine zus.
“Femke zal altijd kwetsbaar en anders zijn. Dat merk ik bij mezelf ook. Ik moest mijn job als zorgkundige opgeven omdat ik scoliose heb, ook een symptoom van 22q11. Als ik op mijn werk zei dat ik moe was, zeiden ze dat ik vast geen zin had om te werken. Ik doe nu administratief werk, telkens interims, maar volg ook een opleiding tot managementassistent. Een knelpuntberoep. Hopelijk vind ik daar een vaste job. Ik heb vaak het gevoel dat we niet genoeg zijn en dat we anders bekeken worden.
“De onduidelijkheid over de toekomst is nog zo’n stressfactor. Ga ik ook psychotische episodes krijgen zoals mijn moeder? Ik heb vrij veel kans om de ziekte van Parkinson te krijgen, maar net zo goed krijg ik die diagnose nooit. Ik zou liever weten wat me nog te wachten staat.”
Tim Osta (44), Ibe (12) en Finn (7)

“Ibe was in de buik al kleiner dan gemiddeld. Volgens UZ Leuven ging het om een niet- opereerbare hartafwijking. Zodra hij geboren werd, zou hij sterven. Mijn vrouw Elke en ik kregen het voorstel om in Leuven te bevallen zodat hij meteen aan de hart-longmachine gekoppeld kon worden. Dat wilden we niet. De natuur mocht beslissen. Elke is opvoeder en ik ben kinesist. We wisten allebei hoe het leven van kinderen met een zware beperking er kan uitzien. Die laatste weken voor de geboorte waren hartverscheurend. We probeerden ons voor te bereiden op een onmogelijk afscheid. Het geboortekaartje vervingen we door een rouwkaartje. Ten laatste drie dagen na de geboorte zou zijn hartje stilvallen. Maar dat is nooit gebeurd. Ibe werd net sterker. De prof in Leuven zag het meteen. ‘Kleine oren die ver van de schedel staan, brede neusbrug, lange vingers en een smal gezicht met kleine ogen: uw kind heeft 22q11.’
“We gingen voor een tweede opinie naar UZ Gent, waar Ibe op drie weken een open hartoperatie onderging om zijn aorta en longslagader van elkaar los te maken. De diagnose van 22q11 werd bevestigd. Hij zou de komende jaren kampen met zware infecties aan de luchtwegen en oren. Omdat hij te weinig calcium aanmaakt, had hij snel een zwak spiergestel. We startten met thuisbegeleiding, kine, logopedie en orthopedagogie. Die baby- en peuterfase was loodzwaar. Telkens moesten we nieuwe oplossingen zoeken voor onbekende problemen. Hij is nu 12. Zijn immuniteit is verbeterd, maar zijn autismespectrumstoornis en mentale beperking nemen de bovenhand. Hij heeft het ontwikkelingsniveau van een vierjarige. We hebben heel ons leven aangepast om Ibe een zo goed als mogelijk leven te geven. Mijn echtgenote werkt ’s avonds op een internaat, ik overdag. Tot zijn 21ste kan hij naar het buitengewoon onderwijs, maar wat daarna? Die vraag hangt als een zwaard van Damocles boven ons hoofd. Sommige kinderen met 22q11 kunnen wel een gewoon leven leiden. Ibe heef het hele pakket gekregen.”
“Ibe is een ‘de novo’, zoals 90 procent van de patiënten. Er is geen erfelijke link. Hij heeft brute pech: een fout in de celdeling. We hebben vijf jaar getwijfeld of we opnieuw zwanger zouden worden. Een tweede kind zou de druk verleggen. We zouden niet alléén maar een kind met een beperking hebben. Finn is tijdens de zwangerschap heel strikt opgevolgd: een vruchtwaterpunctie en elke paar weken een 3D-echo. Hij begint nu op de leeftijd te komen waarop hij fysiek en mentaal sterker wordt dan Ibe. Samenspelen is moeilijk, tenzij bij grof motorische spelletjes. Hij stelt zich sinds kort vragen bij de ontwikkeling van zijn broertje. ‘Waarom roept Ibe zo vaak? Waarom kan hij geen gezelschapsspelletje spelen?’ Ik houd mijn hart al vast voor het moment waarop Ibe beseft dat hij anders is. We willen gewoon dat hij gelukkig is, maar of hij dat ook echt is, zal altijd moeilijk in te schatten zijn.”
Hanna Verlaeckt (39)

“Toen mijn dochter 19 jaar geleden voor de eerste keer aan mijn borst lag, stikte ze bijna in de moedermelk. Ze had een open gehemelte, helemaal achteraan in haar keel. Niemand had het opgemerkt. Ze had bovendien een klompvoetje, wat destijds nog niet zichtbaar was op de prenatale echo’s. Op de afdeling genetica vertelden ze me dat ze 22q11 heeft en dat ik haar die genetische afwijking had doorgegeven. Ik had er nog nooit over gehoord. Niemand uit mijn omgeving trouwens. Die eerste jaren waren rollercoasters van onwetendheid, onbegrip, ontelbaar veel ziekenhuisbezoeken en een slopend schuldgevoel.
“In de lagere school werd ze zwaar gepest omdat ze moeilijk kon praten. Haar huig was te kort om bepaalde klanken te vormen. Ze heeft heel lang logopedie gevolgd en een beugel gedragen om haar huig actiever te maken. In de middelbare school kreeg ze last van migraine. Ze heeft bijna voortdurend hoofdpijn en in lastige situaties begint ze soms te hyperventileren. Ze is nu 19 en ze doet het goed. Ze heeft vrienden bij wie ze terechtkan, ze gaat binnenkort alleen wonen en ze heeft een goede job.
“Mijn dochter heeft net als ik een lichte vorm van 22q11. Ik ben pas beginnen spreken toen ik vier was. Ik kom uit een heel katholiek nest waar ze dat niet zo abnormaal vonden: ‘Ze zal wel praten als ze er klaar voor is.’ Ik werk al een hele tijd halftijds in de zorgsector omdat ik anders geen energie meer heb voor het huishouden. Rond mijn 20ste kreeg ik zwakke polsen en zodra ik 35 werd, werden de spieren in mijn benen zwakker. Ik was vroeger heel sportief, maar nu moet ik blij zijn met wandelingen van 10 kilometer.

“Na onze oudste dochter kregen we nog een dochtertje. We hebben er bewust vijf jaar tussen gelaten. We hebben de NIP-test laten doen en een test om hartproblemen uit te sluiten, maar geen vruchtwaterpunctie om 22q11 op te sporen. We wisten dat er 50 procent kans was dat ze de aandoening ook zou hebben, maar dan was het maar zo. Het zou bijzonder zwaar geweest zijn, maar anderzijds waren we wel al goed voorbereid.
“Ik ben vrijwilliger bij vzw Vecarfa, een platform voor ouders van kinderen met 22q11. Zij voelen zich vaak bijzonder hulpeloos en onwetend. Het syndroom is ongeneeslijk, maar met de juiste zorg kan het wel leefbaar zijn. Meer bekendheid is zo belangrijk. Veel mensen staan er sceptisch tegenover. Ze vragen zich af of er wel echt iets gaande is. Mensen met het syndroom van Down herken je meteen. Met ons lijkt er soms niets aan de hand. Die onwetendheid is enorm frustrerend.”